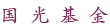南开大学现代光学研究所《Angew. Chem. Int. Ed.》:
---- 绿色合成之单分子水平上的光电协同作用催化自由基偶联反应
1背景介绍
在化学合成领域,构建碳-氮(C–N)键是一项至关重要的反应。传统的合成方法严重依赖钯、铜等贵金属催化剂,不仅成本高昂,还存在催化剂残留、环境污染以及需要预先对反应物进行修饰等问题。虽然科学家们后来发展了自由基偶联策略,但通常仍需要催化剂来启动反应,未能从根本上解决“绿色合成”的难题。
因此,开发一种无需催化剂、反应条件温和、且能在微观尺度上进行精准操控的全新合成方法,成为追求的目标。近年来,定向外电场(OEEF)作为一种“绿色催化剂”崭露头角,为无催化剂合成提供了可能。然而,如何利用电场来驾驭活性极高、寿命极短的自由基,并实现高效的定向偶联,一直是一个巨大的挑战。
针对这一挑战,南开大学现代光学研究所及单分子科学研究中心研究团队结合了定向外电场(OEEF)和原位紫外光照,成功在单分子水平上实现了无需催化剂的C–N自由基偶联反应。

方案1:实现 C-N 偶联的方案对比:(a)传统方法,(b) 本文策略。
2 内容简介
利用紫外光照射反应分子(4,4’-二氨基二苯甲酮),使其羰基生成高活性的自由基。与此同时,利用扫描隧道显微镜裂结技术(STM-BJ)在电极间施加偏压,产生一个强度高达10^9 V/m的定向电场。这个强大的电场能立即“抓住”刚刚产生的自由基,驱动它们发生分子间的氢原子转移(HAT)并最终完成C–N自由基偶联,有效避免了自由基的淬灭或发生副反应。
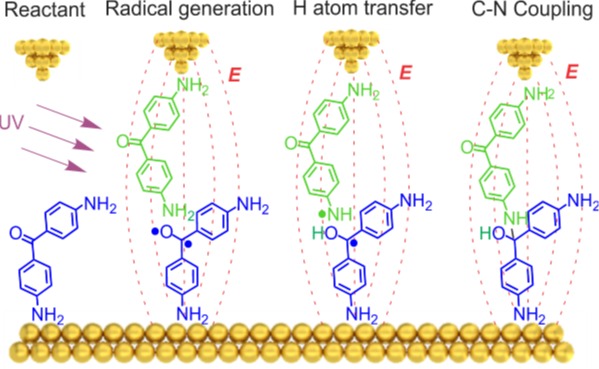
在非极性溶剂TCB中,由于光照和电场的协同作用,一维电导直方图中逐渐出现了一个新的、代表产物分子的低电导 (LC) 峰,如图1b所示,且其强度随光照时间不断增加;与此同时,代表反应物的高电导峰则逐渐降低,直观地展示了反应物向产物的转化。而在极性溶剂PC中,由于电场被屏蔽,该现象完全消失。这一对比实验证明了定向外电场是驱动该反应发生的关键。
图1. 不同溶剂中及不同光照时间下的电导统计图。
电导的二维直方图分析为产物的确定提供了直接而有力的证据。如图2a所示,紫外光激发4,4’-DABP分子激发态,产生碳中心自由基;在电场驱动下,邻近分子的氨基上一个氢原子转移至羰基的氧上,即发生氢原子转移(HAT),产生氮中心自由基;最终,碳自由基与氮自由基发生分子间偶联,形成C–N键。同时二维电导直方图更直观地展示了反应进程:随着紫外光照与电场共同作用时间从1小时延长至3小时,代表产物的LC平台逐渐占据主导地位,而HC平台相应减弱,清晰展现了反应物分子不断转化为更长的产物分子的过程。
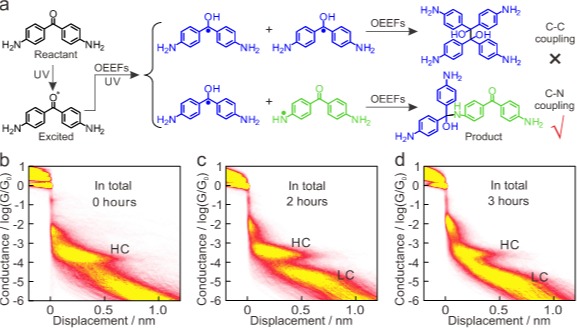
图2. 光电协同催化反应机理及不同光照时间下总的二维电导统计图。
为了更精准地量化反应进程,研究者采用了基于机器学习的聚类分析方法,对海量的电导-位移曲线进行识别和分类。如图3所示,所有曲线主要被分为三类:Cluster A(图3a-c)的特征是仅出现一个HC平台,代表由反应物分子形成的分子结;Cluster B(图3d-f)的特征是先出现一个较短的HC’平台,随后过渡到一个长的LC平台,这被认为由产物分子形成的分子结,其不同平台长度对应于拉伸过程中分子不同锚定基团与电极的连接状态(图3h-i)。定量统计显示,随着反应时间从1小时延长到3小时,代表反应物的Cluster A占比从46.5%显著下降至13.8%,而代表产物的Cluster B占比则从16.8%大幅上升至55.6%。这一数据不仅再次证实了新分子的生成,更为后续反应动力学分析提供了精确的数据基础。
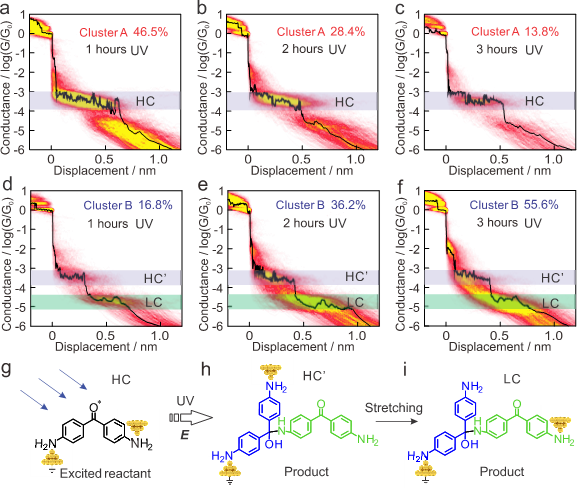
图3. 二维电导统计图的聚类分析及电导平台对应的分子结构。
基于聚类分析得到的产物和反应物比例数据而进行的反应动力学分析发现该反应符合准一级反应模型(图4a)。这与有限元模拟得到的电场分布情况相匹配(图4d),即分子在有限的空间中逐个(或数个)发生反应。反应的进行及其对光电场协同作用的依赖性,通过了多种谱学手段的严格验证。电子顺磁共振波谱(图4b)显示,在紫外光照射下,目标分子溶液产生了明显的自由基信号,且信号强度随照射时间增加。但当仅有光照而无电场时,核磁共振波谱和紫外-可见吸收光谱均未检测到C-N偶联产物的信号,表明单纯光照不足以引发反应。最直接的证据来自高分辨质谱(图4c):对积攒的产物进行分析可发现一个全新的、强度可观的分子离子峰,该质量数恰好与两个4,4’-DABP分子通过一个C–N键偶联并失去一个H原子后的产物分子量精确匹配,确凿地证明了C–N偶联产物的生成。
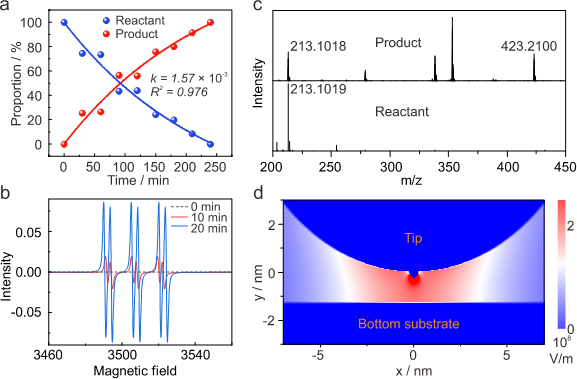
图4. 电场催化反应速率、电子顺磁谱、高分辨率质谱分析、及纳米间隙处电场强度计算分布。
理论计算从能量和电子结构层面深入揭示了电场与光协同催化的微观机制。密度泛函理论(DFT)计算了反应各步骤的能量变化(图5a)。结果表明,定向外电场不仅降低了激发能垒,更重要的是显著降低了整个反应的决速步骤——氢原子转移(HAT)步骤的能垒(TS1):从无电场时的1.30 eV大幅降至0.89 eV。随后的自由基偶联步骤(TS2)的能垒(~0.96 eV)本身较低且受电场影响很小,进一步证实了HAT是反应的控制步骤,与实验得到的准一级动力学模型一致。静电势(ESP)分布(图5b)提供了更直观的解释:无电场时,分子无论基态还是激发态,其ESP分布基本对称;而施加电场后,分子电子云被强烈极化,激发态分子尤其明显,羰基氧原子周围的负电势显著增强,极大地增强了它从相邻分子氨基上夺去氢原子的能力,从而促进了HAT过程。透射谱计算结果与实验观察到的电导演化趋势高度一致,从理论上解释了整个实验现象。
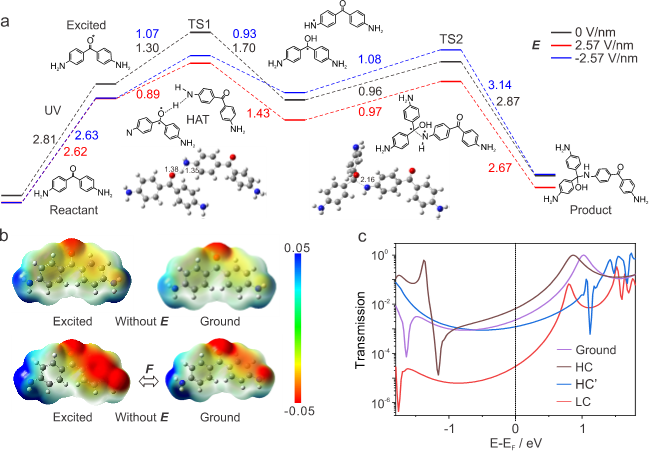
图5. 计算得到的反应过程中能级结构、静电势分布、及电子透射谱。
3 总结概括
本研究通过将定向外电场与紫外光照巧妙协同,在单分子水平成功实现了无需催化剂的C–N自由基偶联反应,利用STM-BJ技术产生的强电场精准驱动光生自由基发生分子间氢原子转移与选择性偶联。结合电导实时监测、EPR与HRMS谱学验证以及理论计算,不仅证实了电场能显著降低反应能垒、促进反应发生,提供了一种绿色合成新范式,为在微观尺度精准操控化学反应路径提供了突破性方法。
4 文章信息
该文章于2025年9月8日以“Catalysis of Radical Coupling Reaction via Synergistic Action of Oriented External Electric Field and Light Irradiation”为题发表于Angewdante Chemie International Edition,文章的第一作者为南开大学现代光学研究所及单分子科学研究中心硕士研究生冯贺发同学。
原文链接:https://onlinelibrary.wiley.com/doi/10.1002/anie.202514789